Le malattie mitocondriali rappresentano un gruppo di disturbi metabolici ereditari che si sviluppano quando i mitocondri dell’organismo non funzionano correttamente. I mitocondri, minuscole strutture cellulari presenti in tutte le cellule ad eccezione dei globuli rossi, sono le centrali energetiche della cellula. Il loro corretto funzionamento è essenziale per la vita di ogni tessuto. Quando i mitocondri non funzionano correttamente, all’interno delle cellule viene generata meno energia. Questo può determinare lesioni cellulari o persino la morte della cellula e un malfunzionamento dei sistemi dell’organismo.
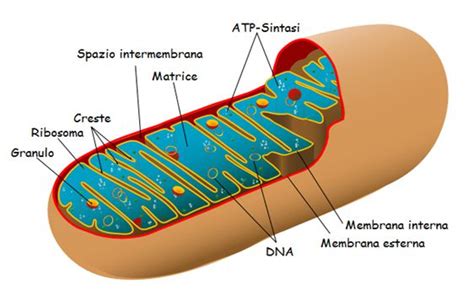
Le malattie mitocondriali possono generare un ampio spettro di sintomi che includono affaticamento cronico, debolezza muscolare, disturbi neurologici e problemi cardiaci. L’eteroplasmia, un fenomeno in cui copie di DNA mutato (Δ-mtDNA) coesistono con copie di DNA non mutato o wild type (wtDNA) nella stessa cellula, è una caratteristica di molte mitocondriopatie legate a mutazioni del DNA mitocondriale (mtDNA).
Origini Genetiche delle Mitocondriopatie
Circa il 75% delle mitocondriopatie è legato a mutazioni del DNA nucleare (nDNA), con il restante 25% dovuto a mutazioni del DNA mitocondriale (mtDNA). Le Delezioni Singole di Larga-Scala del mtDNA (SLSMDs) causano il 10% di tutte le malattie legate a mutazioni del mtDNA. La Sindrome di Pearson (PS), descritta per la prima volta nel 1979, è stata riconosciuta circa 10 anni dopo come appartenente al gruppo delle malattie mitocondriali. La PS è causata da vari tipi di SLSMDs; la più frequente, definita anche "common deletion", ha una lunghezza pari a 4977 bp.
Le malattie ereditarie si verificano quando i genitori trasmettono alla prole i geni difettosi che le causano. A differenza di altre strutture all'interno delle cellule, i mitocondri possiedono materiale genetico proprio, ereditato solo dalla madre. Altro materiale genetico per i mitocondri si trova nel nucleo della cellula, assieme al resto del materiale genetico cellulare, e per essere affetti i bambini devono ereditare un’anomalia da ciascun genitore. La trasmissione materna di malattie neurologiche o di altre condizioni è un fattore importante da considerare.
La Sindrome di Pearson: Manifestazioni Cliniche nell'Infanzia
La Sindrome di Pearson è una malattia grave, spesso letale nei primi anni di vita. La gravidanza e la nascita possono occasionalmente essere complicati da citopenia severa o acidosi metabolica. Un peso neonatale inferiore a 2.500 grammi è riportato nella metà circa dei pazienti. Un ritardo di crescita post-natale, in genere con normali valori di circonferenza cranica, si riscontra nel 70% circa dei casi.
L’anemia, molto frequentemente severa e macrocitica, è pressoché sempre riscontrata al primo emocromo, spesso peraltro in un contesto di pancitopenia. La maggior parte dei pazienti è trasfusione-dipendente già nei primi mesi di vita. Un successivo incremento spontaneo dei valori di emoglobina, con raggiungimento dell’indipendenza trasfusionale, è stato molto spesso segnalato entro i successivi 2-3 anni di vita. Al primo emocromo, piastrinopenia e neutropenia si riscontrano rispettivamente in circa il 50% e il 90% dei pazienti.
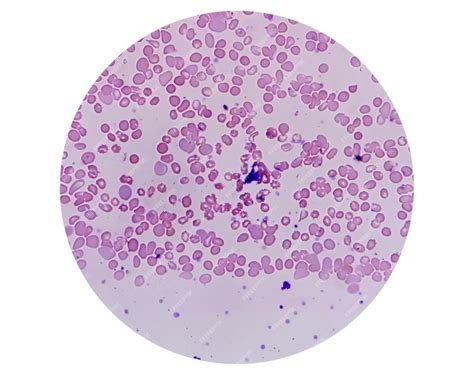
All’aspirato midollare, una riduzione della cellularità con vacuolizzazione dei precursori mieloidi ed eritroidi si apprezza nell’80% circa dei casi. Nell’85% dei pazienti, la colorazione di Pearls mostra la presenza di sideroblasti ad anello.
Coinvolgimento Neurologico e Visivo
Come detto, l’esame neurologico alla nascita è in genere normale, ma anomalie del sistema nervoso compariranno prima o poi in più della metà dei pazienti. I sintomi più frequenti sono ipotonia, ritardo cognitivo, atassia e convulsioni. Una grande varietà di problemi visivi (ptosi, oftalmoplegia, anomalie corneali, cataratta, retinopatia) affliggono circa il 50% dei pazienti.
I disturbi mitocondriali possono causare una vasta gamma di problemi neurologici. Segnali d'allarme che richiedono attenzione immediata sono la presenza di sintomi gravi, la progressione rapida dei disturbi e la comparsa di sintomi in giovane età.
Diagnosi delle Malattie Mitocondriali
Il marker diagnostico per la Sindrome di Pearson è la dimostrazione della presenza del Δ-mtDNA, tramite tecniche di PCR, nei leucociti o in altri tessuti come cute, capelli, fibroblasti, epatociti e cellule di sfaldamento urinario. Un incremento di 1.3-5 volte del lattato nel siero è quasi sempre riscontrato. Visto però che, a volte, i livelli di lattato sono solo lievemente incrementati, un ulteriore strumento diagnostico può essere il rapporto lattato/piruvato, in genere > 20.
All’amminoacidogramma plasmatico, un incremento dei valori di alanina è quasi la regola, ma è possibile anche riscontrare ridotti livelli di citrullina ed arginina. L’elettroencefalogramma segnala talvolta una attività lenta e irregolare, ma è tipicamente alterato soprattutto nei pazienti che stanno progredendo verso la Sindrome di Leigh (KSS).
Per una diagnosi accurata, sono essenziali i test diagnostici: soprattutto l’analisi genetica può rivelare mutazioni nel DNA mitocondriale indicative di malattie mitocondriali ereditarie. I criteri diagnostici per le malattie mitocondriali vengono divisi in maggiori e minori. La presenza di mutazioni del mtDNA o nDNA è un criterio diagnostico molecolare.

Approcci Terapeutici e Gestione del Paziente
La terapia per la Sindrome di Pearson è essenzialmente di supporto. Si basa sulla somministrazione di estratti pancreatici e vitamine liposolubili (A, D, E e K), trasfusioni, cura delle eventuali malattie organo-specifiche associate, degli incidenti metabolici e delle infezioni. Nei pazienti con PS, qualsiasi malattia acuta, causando un incremento delle richieste metaboliche, può accentuare l’acidosi metabolica e portare ad un serio peggioramento delle condizioni generali. Anche per questa ragione, la vaccinazione annuale contro l’influenza deve essere sempre suggerita.
I percorsi terapeutici per le malattie mitocondriali sono una sfida complessa e in continua evoluzione, a causa della vasta gamma di sintomi e della diversità di condizioni. L'attenzione a nutrienti specifici, come coenzima Q10, carnitina e antiossidanti, può favorire la funzione mitocondriale, fornendo un supporto essenziale per migliorare la produzione di energia cellulare. L'esercizio fisico è un componente chiave dello stile di vita per chi affronta malattie mitocondriali.
Associazione Mitocon: ricerca, assistenza e rete scientifica per le malattie mitocondriali
La consulenza genetica è essenziale per le famiglie con una storia di tali disturbi, in quanto offre informazioni sul rischio di trasmissione e sulle opzioni disponibili per la prevenzione. La vita quotidiana per chi affronta una malattia mitocondriale diventa spesso un equilibrio complesso tra la gestione dei sintomi ed il tentativo di mantenere un senso di normalità. Il supporto ai pazienti è fondamentale in questo contesto: essere parte di gruppi di aiuto o comunità consente di condividere esperienze, ricevere consigli pratici e trovare conforto nella comprensione reciproca. La consulenza psicologica è un'importante risorsa per affrontare gli impatti emotivi della malattia.
Prognosi e Ricerca
Le più frequenti cause di morte nella Sindrome di Pearson sono episodi di acidosi metabolica intrattabile, sepsi e, meno frequentemente, insufficienza epatica o renale. È verosimile che il miglioramento della sopravvivenza notato in anni più recenti sia legato essenzialmente al miglioramento generale delle terapie di supporto. La ricerca sulle malattie mitocondriali ha conosciuto significativi avanzamenti negli ultimi anni, alimentando speranze di potenziali cure e terapie future.
Casi Clinici Esemplificativi
Un esempio di come la ricerca e la diagnosi precoce possano fare la differenza è il caso di un bambino nato pretermine a 31 settimane, con un peso di 1 chilo e 400 grammi, dopo un parto cesareo d’urgenza dovuto al liquido amniotico in eccesso. Nonostante analisi chimiche, indagini strumentali e metaboliche negative, il sospetto di una patologia mitocondriale ha portato all’avvio urgente di un sequenziamento dell’esoma. La diagnosi è stata encefalomiopatia mitocondriale molto rara, nota come sindrome da deplezione del DNA mitocondriale tipo 13, caratterizzata da ipotonia, difficoltà di suzione, encefalopatia con severo ritardo dello sviluppo e persistente acidosi lattica. Grazie a questa diagnosi, l’approccio terapeutico è stato mirato, comprendendo come i mitocondri del paziente venissero precocemente distrutti dalla cellula a causa della mutazione genetica. Il bambino ha iniziato a crescere, i valori di lattato e glicemia sono tornati normali e ha raggiunto le 38 settimane post-concezionali.
Un altro esempio è il caso di Anna, una bambina di 5 anni affetta da una malattia mitocondriale che le ha tolto la vista ma affinato l’udito, dimostrando come la vita possa essere ricca di esperienze sensoriali alternative. Sophie Teresa, invece, è uno dei dieci casi conosciuti al mondo di mutazione del gene FDX2, una malattia mitocondriale rarissima, la cui diagnosi è stata possibile grazie a Telethon, ponendo speranze nella ricerca. La collaborazione tra Fondazione Cariplo e Fondazione Telethon, attraverso la selezione di 21 nuovi progetti di ricerca di base per un totale di circa 5 milioni di euro, evidenzia l'impegno nella comprensione di queste complesse patologie.
Altri Disturbi Mitocondriali
Oltre alla Sindrome di Pearson, esistono altre patologie mitocondriali con manifestazioni diverse:
- Neuropatia Ottica Ereditaria di Leber (LHON): Provoca la perdita progressiva della vista da entrambi gli occhi, talvolta associata a problemi cardiaci o muscolari. I sintomi iniziano spesso tra i 20 e i 40 anni.
- Sindrome di Leigh: Solitamente inizia durante il primo anno di vita, con sintomi come problemi di alimentazione, scarso controllo della testa, ritardi dello sviluppo, vomito, irritabilità, pianto continuo e convulsioni. Possono verificarsi episodi di acidosi lattica che portano a problemi respiratori e renali. Alcuni bambini con un tipo specifico di sindrome di Leigh traggono beneficio da integratori a base di tiamina (vitamina B1) e biotina.
- Encefalomiopatia Mitocondriale, Acidosi Lattica ed Episodi Simili all’Ictus (MELAS): I bambini affetti possono presentare debolezza e dolore muscolare, cefalea ricorrente, perdita dell’udito, inappetenza, vomito e convulsioni. La maggior parte dei pazienti colpiti presenta episodi simili all’ictus che iniziano prima dei 40 anni di età, con conseguenti danni cerebrali progressivi.
Questi esempi sottolineano la complessità e la variabilità delle malattie mitocondriali, rendendo la ricerca e la diagnosi tempestiva pilastri fondamentali per la gestione e il miglioramento della qualità di vita dei pazienti.
tags: #mitocondriale #infantile #fetale