Le malattie rare rappresentano una vasta e complessa area della medicina, caratterizzate dalla loro bassa prevalenza nella popolazione generale, ma che, sommate, colpiscono un numero considerevole di individui. Queste patologie, spesso di origine genetica, possono manifestarsi fin dalla nascita, presentando sfide significative sia nella diagnosi che nel trattamento. Comprendere le cause sottostanti, riconoscere i sintomi e conoscere le opzioni terapeutiche disponibili è fondamentale per migliorare la qualità della vita dei pazienti e delle loro famiglie.
La Natura Genetica delle Malattie Rare Congenite
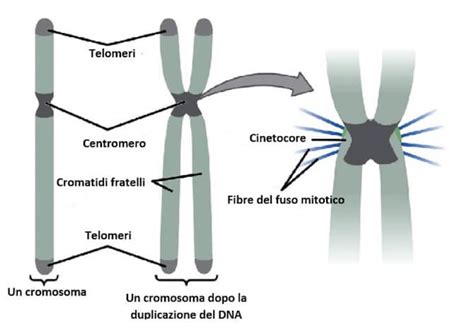
Molte delle malattie che si manifestano fin dalla nascita hanno una base genetica. Il DNA, contenuto in ogni cellula del nostro corpo, racchiude le informazioni ereditarie che vengono trasmesse dai genitori ai figli. Questo DNA è organizzato in 23 coppie di cromosomi, per un totale di 46. Ciascuna coppia è composta da un cromosoma ereditato dalla madre e uno dal padre. All'interno di questi cromosomi si trovano i geni, circa 24.000 nell'uomo, che determinano le nostre caratteristiche e le funzioni del nostro organismo.
Le anomalie genetiche possono manifestarsi in diversi modi. Alcune malattie sono causate da mutazioni in un singolo gene, dando origine alle cosiddette malattie monogeniche. Queste possono essere a trasmissione autosomica dominante, recessiva o legata al cromosoma X. Nelle malattie autosomiche dominanti, l'alterazione di una sola copia di un gene è sufficiente a causare la malattia, poiché il gene mutato "domina" su quello normale. Tuttavia, in alcuni casi, la mutazione può verificarsi "de novo", ovvero spontaneamente durante la formazione delle cellule riproduttive o nelle prime fasi dello sviluppo embrionale, piuttosto che essere ereditata da un genitore. Un fenomeno interessante nelle malattie autosomiche dominanti è la "penetranza incompleta", dove individui portatori della mutazione genetica non manifestano la malattia o la presentano in forma molto attenuata, rendendo a volte difficile la diagnosi familiare e la predizione del rischio ereditario.
Nelle malattie autosomiche recessive, invece, è necessario che entrambe le copie di un gene siano alterate per manifestare la patologia. I genitori, in questo caso, sono portatori sani, possedendo una copia mutata e una copia normale del gene, e non presentano sintomi. Il rischio di avere un figlio affetto da una malattia autosomica recessiva è del 25% per ogni gravidanza in una coppia di portatori sani.
Le malattie genetiche legate al cromosoma X, come molte forme di distrofia muscolare o emofilia, seguono un pattern di trasmissione specifico. Se un gene mutato si trova sul cromosoma X, le donne, avendo due cromosomi X, possono compensare l'alterazione con la copia sana del gene presente sull'altro cromosoma X, risultando portatrici sane o manifestando forme più lievi. Gli uomini, avendo un solo cromosoma X, manifestano la malattia se ereditano la copia mutata.
Oltre alle mutazioni geniche singole, le malattie genetiche possono derivare da alterazioni cromosomiche, come la presenza di un cromosoma in più (trisomia), un cromosoma in meno (monosomia) o riarrangiamenti strutturali. La Sindrome di Down (Trisomia 21), ad esempio, è causata dalla presenza di un cromosoma 21 in più.
Infine, esistono difetti alla nascita multifattoriali, causati da una combinazione complessa di fattori genetici e ambientali. La prevenzione di questi difetti spesso include uno stile di vita sano, l'assunzione di acido folico prima e durante la gravidanza, e la vaccinazione (ad esempio contro la rosolia).
Esempi Specifici di Malattie Rare Congenite
L'ampio spettro delle malattie rare congenite include patologie con manifestazioni molto diverse.
La Sindrome di DiGeorge, nota anche come Sindrome da delezione 22q11.2, è una malattia rara che colpisce circa 1 persona su 4.000. Nella maggioranza dei casi, non è ereditaria, ma in una percentuale del 10-20% il cromosoma 22 difettoso viene trasmesso da un genitore con un meccanismo autosomico dominante. La diagnosi avviene solitamente subito dopo la nascita tramite un esame del sangue che rileva il difetto genetico. Questa sindrome è inserita nell'elenco delle patologie rare esenti dal costo del ticket, con codice RCG160, afferente al gruppo delle Immunodeficienze primarie. È studiata a livello europeo all'interno di ERN Rita.
La Sindrome di Prader-Willi (PWS) è un'altra rara patologia genetica congenita, che colpisce entrambi i sessi con la stessa frequenza, con una prevalenza stimata di 1 caso su 25.000. La PWS è causata da un'anomalia genetica sul braccio lungo del cromosoma 15 ereditato dal padre. Questa anomalia può manifestarsi con geni paterni assenti o difettosi, o con la presenza di due copie del cromosoma 15 di origine materna. L'alterazione genetica influisce sulla funzione dell'ipotalamo, una ghiandola cerebrale cruciale per la regolazione della crescita psico-fisica, dello sviluppo sessuale, della temperatura corporea, del senso di sazietà, del sonno e dell'umore. La sindrome è perlopiù causata da un errore genetico casuale e non è ereditaria. La diagnosi si basa sull'osservazione clinica e viene confermata da analisi genetiche.
I sintomi della Sindrome di Prader-Willi variano notevolmente tra i pazienti e nel corso della vita, ma tipicamente si presentano in due fasi. Nel periodo neonatale e nella prima infanzia, si osserva una marcata riduzione del tono muscolare (ipotonia), che causa difficoltà di suzione, scarso accrescimento e ritardo nello sviluppo neuromotorio. Successivamente, tra i due e i quattro anni, l'ipotonia migliora e subentra un aumento patologico dell'appetito (iperfagia) dovuto alla mancanza del senso di sazietà. Senza un adeguato controllo calorico, si sviluppa un'obesità grave, resistente ai trattamenti, che porta a complicazioni cardiorespiratorie, metaboliche, epatiche e osteoarticolari. Altre manifestazioni includono disfunzioni ormonali (ipogonadismo, ipotiroidismo, bassa statura, iposurrenalismo), dismorfismi facciali, mani e piedi piccoli, ipopigmentazione cutanea, ridotta sensibilità al dolore, patologie oculistiche, disabilità intellettiva (lieve o moderata) e disturbi comportamentali e psichiatrici. L'impatto sulla vita del paziente è significativo, limitando le relazioni sociali e l'indipendenza, ma con un adeguato supporto, è possibile un buon inserimento scolastico e lavorativo. L'obesità è il principale fattore che influenza la prognosi.

La Fibrosi Cistica (FC) è un'altra malattia genetica autosomica recessiva, causata da mutazioni nel gene CFTR. In Italia, colpisce circa 1 neonato su 3.000, indistintamente maschi e femmine. Le persone con una copia normale e una mutata del gene CFTR sono portatori sani, senza manifestazioni cliniche. Il test per l'identificazione del portatore è offerto dal Servizio Sanitario Nazionale in specifiche circostanze. In una coppia di portatori, il rischio di avere un figlio affetto da FC è del 25% ad ogni gravidanza. I sintomi includono infezioni respiratorie ricorrenti e croniche, cattiva digestione, malnutrizione, ritardo di crescita, epatopatia cronica, aumento del sale nel sudore e infertilità maschile. Lo screening neonatale obbligatorio permette la diagnosi precoce nella maggior parte dei casi.
I trattamenti per la fibrosi cistica si concentrano sulla gestione dei sintomi: antibiotici per le infezioni, farmaci per fluidificare le secrezioni, fisioterapia respiratoria, dieta ipercalorica e, nei casi più gravi, trapianto polmonare. Farmaci modulatori del gene CFTR sono disponibili per specifiche mutazioni, migliorando la funzione della proteina difettosa e riducendo i sintomi, ma non sono efficaci per tutte le forme della malattia. L'aspettativa di vita per i pazienti con FC è aumentata significativamente grazie ai progressi terapeutici, con oltre il 50% della popolazione affetta che oggi è costituita da adulti, e un'aspettativa di vita che si aggira attorno ai 40 anni.

Diagnosi e Approcci Terapeutici
La diagnosi precoce è un pilastro fondamentale nella gestione delle malattie rare congenite. In molti casi, come per la Sindrome di DiGeorge e la Fibrosi Cistica, lo screening neonatale obbligatorio permette di identificare la patologia subito dopo la nascita. Per la Sindrome di Prader-Willi, la diagnosi si basa sull'osservazione clinica dei segni e sintomi caratteristici, seguita da analisi genetiche molecolari o citogenetiche mediante un semplice prelievo di sangue.
La presa in carico multidisciplinare è essenziale per affrontare la complessità di queste patologie. Un team di specialisti, che può includere pediatri, internisti, endocrinologi, genetisti, neuropsichiatri, cardiologi, pneumologi, fisioterapisti, nutrizionisti e psicologi, lavora in sinergia per definire un percorso di cura personalizzato.
Le terapie disponibili mirano a gestire i sintomi, migliorare la qualità della vita e, quando possibile, correggere le anomalie sottostanti. Per la Sindrome di Prader-Willi, ad esempio, la terapia con ormone della crescita (GH) favorisce una crescita adeguata e migliora la composizione corporea, mentre la terapia ormonale sostitutiva con steroidi sessuali è importante per indurre la pubertà e prevenire l'osteoporosi. La fisioterapia è cruciale per migliorare le capacità motorie e correggere le problematiche posturali, mentre la psicoterapia aiuta a gestire i disturbi comportamentali.
Per le malattie genetiche monogeniche come la fibrosi cistica, oltre ai trattamenti sintomatici, sono in fase di sviluppo e applicazione terapie innovative, come i farmaci modulatori del gene CFTR, che agiscono direttamente sulla causa molecolare della malattia. Sebbene la terapia genica sia ancora in fase di ricerca per molte di queste patologie, i progressi nel campo della genetica e della biologia molecolare aprono prospettive promettenti per il futuro.
Incontro con Domenica Taruscio - Malattie genetiche rare
Informazione e Supporto
La complessità delle malattie rare rende cruciale l'accesso a informazioni accurate e aggiornate. In Italia, il Telefono Verde Malattie Rare (TVMR) 800.89.69.49 offre supporto e indicazioni sui centri specializzati e le associazioni di pazienti. A livello europeo, reti come ERN Rita facilitano la collaborazione tra centri esperti per la diagnosi e il trattamento dei disordini immunologici rari.
Le associazioni di pazienti svolgono un ruolo vitale nel fornire supporto psicologico, informazioni pratiche e nel promuovere la ricerca e la sensibilizzazione su queste patologie. Organizzazioni come la Lega Italiana Fibrosi Cistica sono fondamentali nel migliorare l'assistenza sanitaria, tutelare i diritti e promuovere una migliore qualità della vita per le persone affette da fibrosi cistica.
La comprensione delle cause genetiche, la diagnosi tempestiva, un approccio terapeutico multidisciplinare e il supporto delle reti informative e associative sono elementi chiave per affrontare le sfide poste dalle malattie rare congenite, migliorando significativamente la vita dei pazienti e delle loro famiglie.