Le displasie scheletriche fetali rappresentano un gruppo eterogeneo di patologie caratterizzate da anomalie nello sviluppo e nella crescita del tessuto osteocartilagineo. Queste condizioni, la cui classificazione è in continua evoluzione grazie ai progressi nella ricerca genetica e diagnostica, possono manifestarsi con un'ampia gamma di gravità, spaziando da forme letali in epoca prenatale o neonatale a condizioni compatibili con la vita, sebbene spesso associate a bassa statura disarmonica e altre complicanze. La diagnosi prenatale assume un ruolo cruciale nella gestione di queste patologie, permettendo un counseling genetico mirato e la pianificazione di interventi terapeutici o palliativi.
La Genesi delle Displasie Scheletriche: Anomalie Genetiche e Sviluppo Osseo
Le displasie scheletriche sono, nella maggior parte dei casi, il risultato di mutazioni genetiche che colpiscono geni coinvolti in complessi pathways di segnalazione essenziali per la corretta formazione e crescita dello scheletro. Questi pathways regolano processi fondamentali come la proliferazione e la differenziazione dei condrociti nella cartilagine di accrescimento, la sintesi delle proteine della matrice extracellulare e l'ossificazione encondrale.
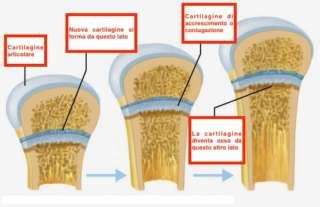
L'ossificazione encondrale, il processo attraverso cui si sviluppano le ossa lunghe, le vertebre e le costole, è particolarmente sensibile alle alterazioni genetiche. In questa modalità di ossificazione, le cellule mesenchimali progenitrici si differenziano in condrociti che formano un modello cartilagineo, successivamente sostituito da tessuto osseo. La cartilagine di accrescimento (Growth Plate, GP) è una zona dinamica dove si alternano zone di riserva, proliferazione, pre-ipertrofia e ipertrofia dei condrociti. Numerosi pathways di segnale, tra cui Indian Hedgehog (IHH) e PTHrP, BMPs, WNTs, Notch, CNP/NPR2 e FGFs, giocano un ruolo fondamentale nel regolare questo complesso processo.
Le mutazioni possono interessare geni coinvolti in questi pathways di segnale o geni che codificano per le proteine della matrice extracellulare. Ad esempio, mutazioni nel gene FGFR3 (Recettore del fattore di crescita dei fibroblasti 3) sono responsabili di forme gravi come la displasia tanatoforica. Questo gene regola la crescita e lo sviluppo osseo, e le sue mutazioni inattivano vie di segnalazione che inibiscono la crescita della cartilagine e dell'osso. Allo stesso modo, anomalie nei geni del pathway WNT possono causare sindromi come la sindrome di Robinow.
Un ruolo centrale è ricoperto anche dalle proteine della matrice extracellulare, come il collagene. Il collagene di tipo II, ad esempio, è fondamentale per la formazione della cartilagine e le sue alterazioni, dovute a mutazioni nel gene COL2A1, possono portare a displasie scheletriche. La mutazione nel gene COL2A1 sul cromosoma 12 è specifica per la displasia spondiloepifisaria congenita, una condizione che spesso si associa a problemi visivi e uditivi.
Classificazione e Caratteristiche delle Displasie Scheletriche Fetali
Le displasie scheletriche vengono classificate in base a criteri clinici, radiografici e molecolari. L'International Skeletal Dysplasias Society (ISDS) ha revisionato la nosologia, identificando centinaia di condizioni raggruppate in decine di gruppi. La gravità di queste patologie varia considerevolmente:
Displasie Scheletriche Letali
Queste forme sono incompatibili con la vita o comportano una sopravvivenza molto limitata.
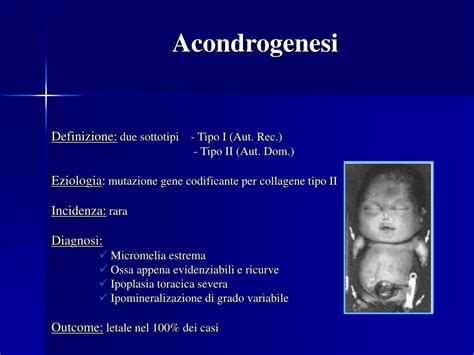
- Acondrogenesi: Caratterizzata da grave ipomineralizzazione, scarsa ossificazione dei corpi vertebrali e assenza di sacro. Può presentare polidramnios e idrope fetale.
- Displasia Tanatoforica (TD): Una delle forme più gravi, caratterizzata da grave bassa statura disarmonica, arti estremamente corti, torace stretto e deformità vertebrali. Si distinguono due tipi principali:
- TD di tipo 1: Femori incurvati a "cornetta del telefono".
- TD di tipo 2: Cranio a trifoglio, più grave della TD1.I segni morfologici caratteristici visibili con l'ecografia prenatale includono accorciamento delle ossa lunghe, translucenza nucale aumentata nel primo trimestre, deficit di crescita marcato, macrocefalia relativa, torace stretto e appiattimento dei corpi vertebrali nel secondo trimestre. Si associa spesso a polidramnios. La trasmissione è autosomica dominante, ma la maggior parte dei casi deriva da mutazioni de novo nel gene FGFR3.
- Osteogenesi Imperfetta tipo II: Una forma letale di osteogenesi imperfetta, caratterizzata da fragilità ossea estrema, numerose fratture fetali, ipomineralizzazione severa e costole corte e fratturate.
- Displasia Scheletrica di tipo Greenberg: Una displasia scheletrica estremamente rara e letale, associata ad anomalie nella biosintesi del colesterolo, causata da mutazioni nel gene LBR. Si manifesta con idrope fetale, nanismo grave con arti corti, anomalie del viso, ipoplasia polmonare, polidattilia e igroma cistico. La diagnosi può essere supportata dall'analisi del profilo degli steroli.
- Sindrome delle coste corte Polidattilia (es. Sindrome di Jeune, Sindrome di Saldino-Noonan): Caratterizzata da torace stretto e ipoplasico con coste corte, spesso associata a polidattilia e altre anomalie viscerali.
Displasie Scheletriche Moderate e Lievi
Queste forme sono generalmente compatibili con la vita, sebbene possano richiedere interventi medici e chirurgici specifici.
- Acondroplasia: La più comune displasia scheletrica, causata da mutazioni nel gene FGFR3. È caratterizzata da bassa statura disarmonica con arti sproporzionatamente corti rispetto al tronco, macrocefalia relativa e facies caratteristica. La diagnosi ecografica può essere difficile nel secondo trimestre, diventando più evidente nel terzo.
- Ipocondroplasia: Una forma più lieve di acondroplasia, anch'essa legata a mutazioni nel gene FGFR3. Causa bassa statura non grave con sproporzione tra tronco e arti.
- Displasia Campomelia: Caratterizzata da incurvamento delle ossa lunghe, in particolare femore e tibia. Può essere associata a varie anomalie genetiche e cromosomiche. La presenza di curvature patologiche con displasia moderata o lieve deve far pensare a campomelia.
- Displasia Spondiloepifisaria Congenita (SEDc): Associata a mutazioni nel gene COL2A1, causa problemi alla vista e all'udito. Si manifesta con bassa statura, tronco corto, anomalie delle vertebre e delle epifisi.
L'Importanza dello Studio Ecografico Fetale
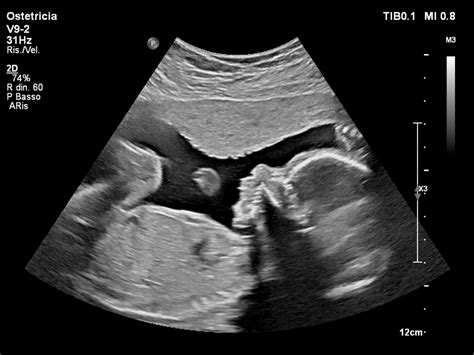
Lo studio ecografico dello scheletro fetale è uno strumento diagnostico fondamentale per l'identificazione delle displasie scheletriche. L'esame è eseguibile agevolmente dalla 13a alla 30a settimana di gestazione, diventando progressivamente più difficile in epoche successive. Una valutazione accurata richiede esperienza dell'ecografista e l'uso di apparecchiature adeguate.
L'ecografia permette di valutare:
- Biometria degli arti: Misurazione della lunghezza delle ossa lunghe (femore, omero, tibia, perone, radio, ulna) e loro segmenti: rizomelico (femore/omero), mesomelico (tibia/perone, radio/ulna) e acromelico (mano/piede). Valori inferiori al 5° percentile per l'epoca gestazionale possono indicare un accorciamento.
- Morfologia degli arti: Presenza di curvature patologiche, fratture, mineralizzazione anomala. L'alterazione dell'asse tra segmento mesomelico e acromelico può definire deformità come il piede torto.
- Anomalie delle dita: Sindattilia (fusione delle dita), polidattilia (dita sovrannumerarie), clinodattilia (deviazione laterale), ectrodattilia (assenza di dita) e camptodattilia (flessione delle articolazioni interfalangee).
- Torace: Biometria (ipoplasia, torace stretto) e morfologia delle coste (corte, ricurve, fratturate). Un torace eccessivamente piccolo è un importante indice di ipoplasia polmonare e prognosi infausta.
- Cranio e Faccia: Biometria (macrocefalia, microcefalia), bozze frontali, radice del naso infossata, schisi labiale/palato, iper/ipotelorismo, cranio a trifoglio, micrognazia. La macrocefalia è presente in acondroplasia e displasia tanatoforica.
- Colonna vertebrale: Valutazione della mineralizzazione e della morfologia dei corpi vertebrali (es. platispondilia).
- Altri organi e apparati: Studio del cuore fetale, valutazione di polidramnios, idrope fetale, anomalie renali o cerebrali.
Diagnosi Differenziale e Integrazione con Analisi Molecolari
La diagnosi ecografica delle displasie scheletriche può essere complessa a causa dell'elevata variabilità fenotipica e della sovrapposizione di quadri clinici. In presenza di un sospetto ecografico, è fondamentale procedere con un'accurata descrizione dei segni riscontrati e considerare una consulenza genetica.
L'integrazione con dati molecolari è spesso imprescindibile per una diagnosi precisa e per fornire un adeguato counseling genetico. L'analisi molecolare prenatale, tramite villocentesi o amniocentesi, può confermare la diagnosi dimostrando la mutazione genetica specifica. L'analisi dell'esoma (Whole Exome Sequencing, WES) sta emergendo come uno strumento potente per diagnosticare una vasta gamma di displasie scheletriche, anche in casi di bassa statura idiopatica.
Studi recenti hanno dimostrato che l'analisi prenatale dell'esoma può individuare l'81% dei casi di displasia scheletrica, offrendo un'ulteriore risorsa diagnostica. L'analisi retrospettiva di casi afferiti a unità di diagnosi prenatale ha evidenziato come, nonostante i progressi dell'ecografia, l'approfondimento molecolare e autoptico rimangano essenziali.
Gestione Clinica e Aspetti Prognostici
La gestione delle displasie scheletriche fetali dipende dalla gravità della condizione:
- Forme Letali: In questi casi, la decisione riguardo all'interruzione volontaria di gravidanza (IVG) è una scelta complessa che richiede un counseling genetico approfondito. Se la gravidanza prosegue, la gestione si concentra sulla prevenzione di complicanze durante la gravidanza e il parto.
- Forme Compatibili con la Vita: Il trattamento si concentra sulla gestione dei sintomi e delle complicanze. I bambini nati con gravi deformità toraciche possono sviluppare difficoltà respiratorie che richiedono cure intensive neonatali. La sostituzione di articolazioni, come quella dell'anca, può essere necessaria in età pediatrica o adulta. La valutazione anestesiologica pre-operatoria è fondamentale. Problemi della vista e dell'udito, frequenti in alcune displasie come la spondiloepifisaria congenita, richiedono test audiometrici ed esami oculistici specialistici.
La prognosi varia ampiamente. Le forme letali comportano il decesso in utero o poco dopo la nascita. Le forme non letali, sebbene possano comportare disabilità significative, permettono una vita con adeguate cure mediche e supporto.
Displasia Congenita dell'Anca
Considerazioni sulla Diagnosi e sul Counseling Genetico
La diagnosi prenatale delle displasie scheletriche è un processo in continua evoluzione. Sebbene l'ecografia rimanga uno strumento primario, la sua accuratezza è influenzata dall'epoca gestazionale, dall'esperienza dell'operatore e dalla complessità del quadro clinico. L'integrazione con tecniche di imaging avanzate e, soprattutto, con l'analisi genetica è fondamentale per una diagnosi definitiva.
Il counseling genetico gioca un ruolo insostituibile nel fornire ai genitori informazioni dettagliate sulla patologia, sul rischio di ricorrenza e sulle opzioni disponibili. La comprensione della trasmissione ereditaria, sia essa autosomica dominante, recessiva o legata a mutazioni de novo, è cruciale per una corretta valutazione del rischio.
Le displasie scheletriche fetali rappresentano una sfida diagnostica e terapeutica significativa. Tuttavia, i continui progressi nella genetica, nella diagnostica per immagini e nelle terapie mediche e chirurgiche offrono speranza per una migliore gestione e un miglioramento della qualità della vita per i bambini affetti da queste complesse condizioni.
tags: #displasia #scheletrica #fetale #tipo